
Recently, Nature Reviews Cancer published an in-depth review that comprehensively analyzes the latest advances, underlying mechanisms, and future translational directions of context-dependent synthetic lethality. Below is a structured summary of this important article.
I. What Is Synthetic Lethality?
The concept of synthetic lethality is elegant: it exploits unique functional vulnerabilities that arise in cancer cells due to specific genetic alterations or cellular states—vulnerabilities that are absent in normal cells. In simple terms, when either condition A or condition B occurs alone, the cell survives; but when both A and B are lost simultaneously, the cell dies.
The power of this strategy lies in its ability to selectively target cancer cells while sparing healthy tissue. So, what types of “genetic contexts” can create these lethal vulnerabilities? The review highlights four major categories:
- Direct oncogene activation: Activation of oncogenic pathways not only drives tumor growth but also creates “addiction” to those pathways. For example, cancers with BRAF mutations become highly dependent on downstream MEK and ERK signaling nodes, creating opportunities for synthetic lethality.
- Loss of tumor suppressor genes: This is the most clinically successful area to date. A classic example is mutations in BRCA1/2, which cause homologous recombination deficiency (HRD). These cancer cells become dependent on PARP enzymes for survival. PARP inhibitors exploit this synthetic lethal interaction and have achieved major clinical success.
- Passenger deletions: During tumor evolution, deletion of key tumor suppressor genes can also remove neighboring genes. For instance, loss of CDKN2A is often accompanied by deletion of the nearby MTAP gene, creating a therapeutic window for targeting PRMT5 or MAT2A.
- Genomic instability: Tumors with microsatellite instability (MSI-H), due to mismatch repair defects, form abnormal DNA structures that require WRN helicase for resolution. Inhibiting WRN leads to DNA fragmentation and cancer cell death.
II. Core Mechanisms Underlying Synthetic Lethality
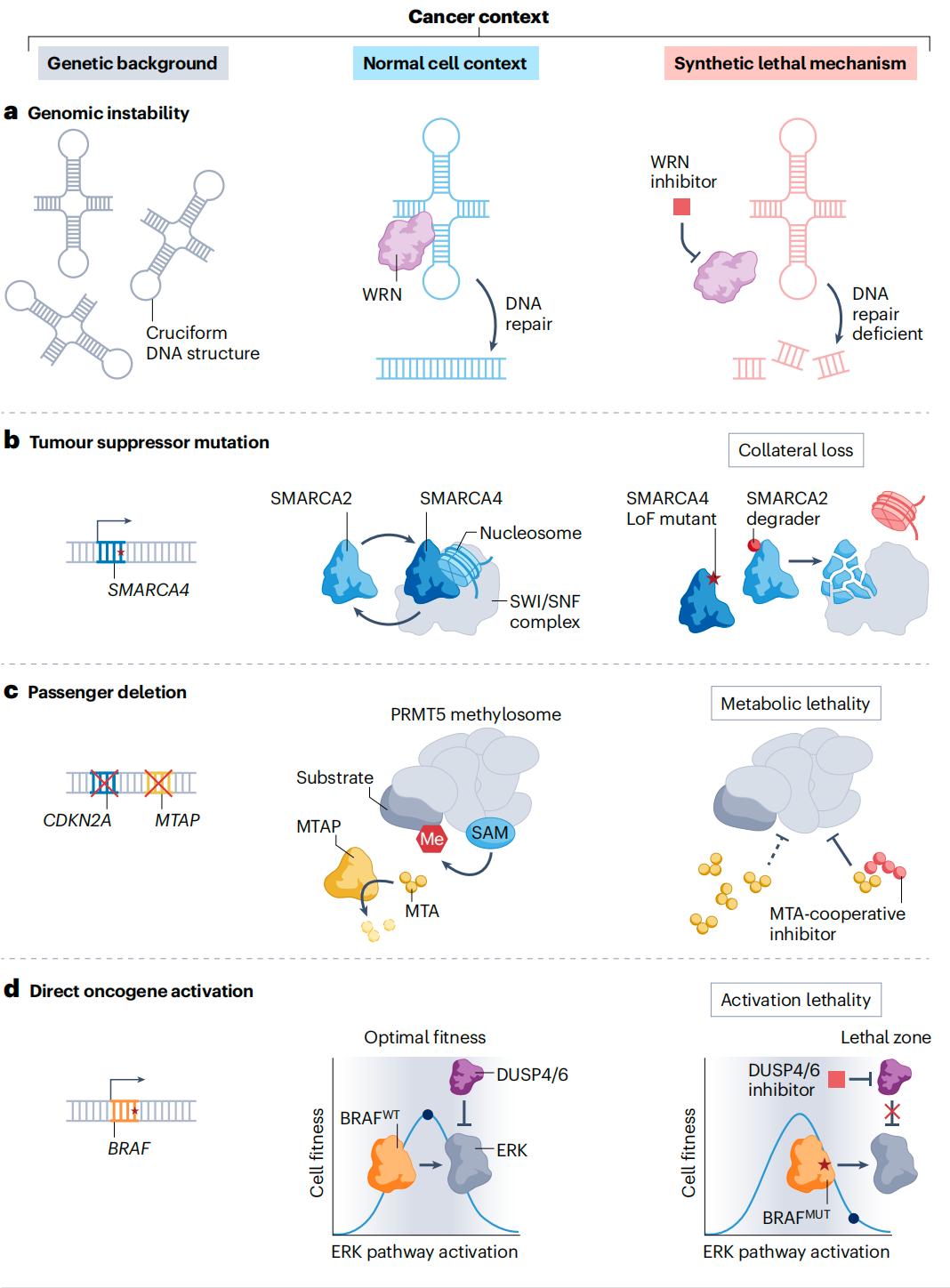
A deep understanding of molecular mechanisms is essential for guiding drug design. Several common modes of action have been identified:
- DNA repair deficiencies: As seen with PARP and WRN, many cancers harbor defects in DNA repair pathways and rely on alternative mechanisms for survival.
- Paralog dependency (redundant essential genes): Normal cells often possess paralogous genes that compensate for each other. When cancer cells lose one (e.g., SMARCA4 in the SWI/SNF complex), the remaining paralog (SMARCA2) becomes essential. Targeting SMARCA2 can selectively kill these cancer cells.
- Metabolic lethality: Loss of MTAP leads to accumulation of the metabolite MTA, which partially inhibits PRMT5. As a result, MTAP-deficient cells become hypersensitive to PRMT5 inhibition.
- Activation lethality: Synthetic lethality does not always require inhibition. In tumors with BRAF or NRAS mutations, inhibiting negative feedback regulators (such as DUSP4/6) can cause hyperactivation of the ERK pathway. This “signal overload” can be toxic to cancer cells.
III. Key Targets and Clinical Development Landscape
To better illustrate the current R&D landscape, the review summarizes representative synthetic lethality targets and their clinical status. Beyond the already successful PARP inhibitors, drugs targeting PRMT5, WRN, CDK2, and others are advancing rapidly through clinical development.
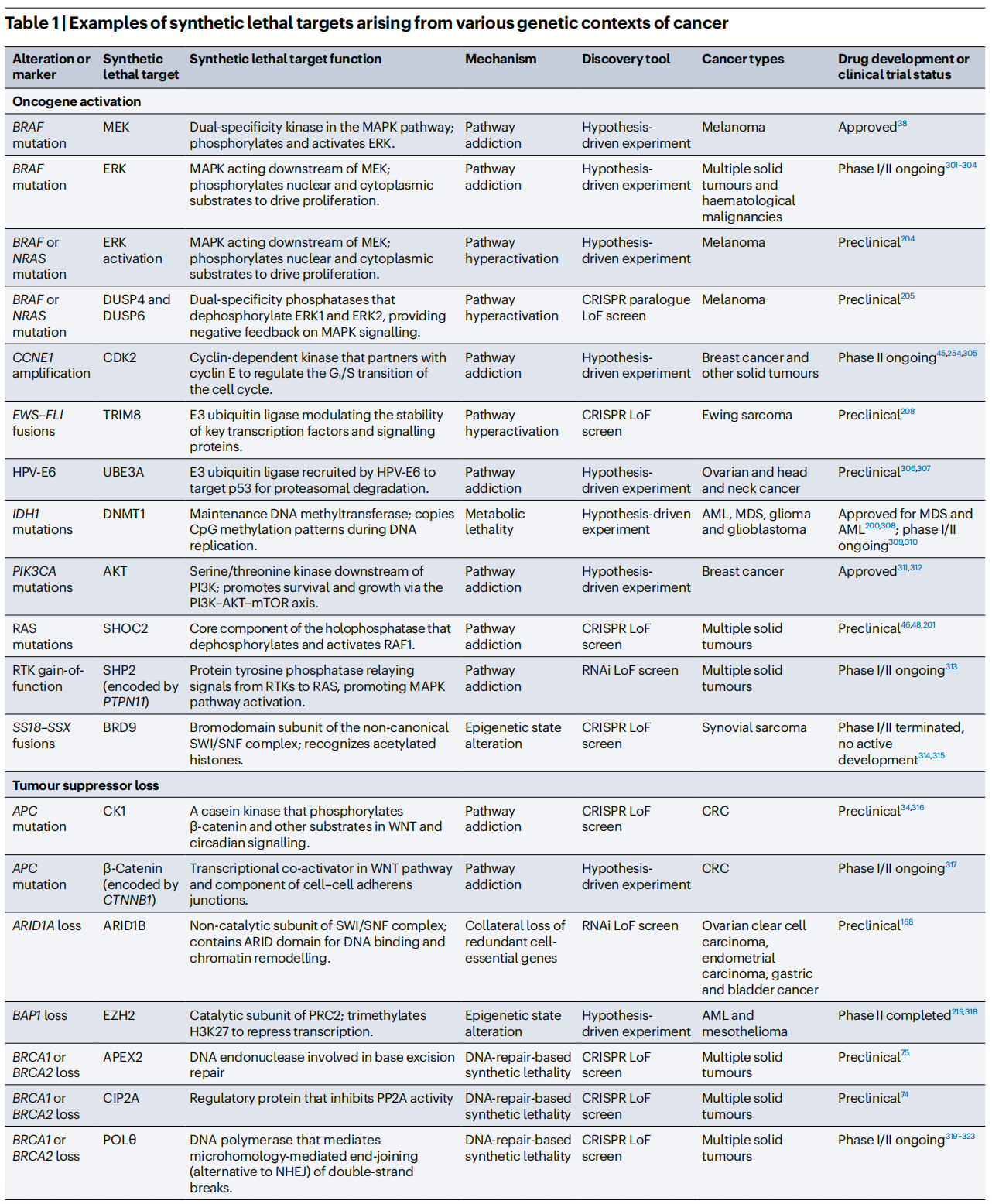
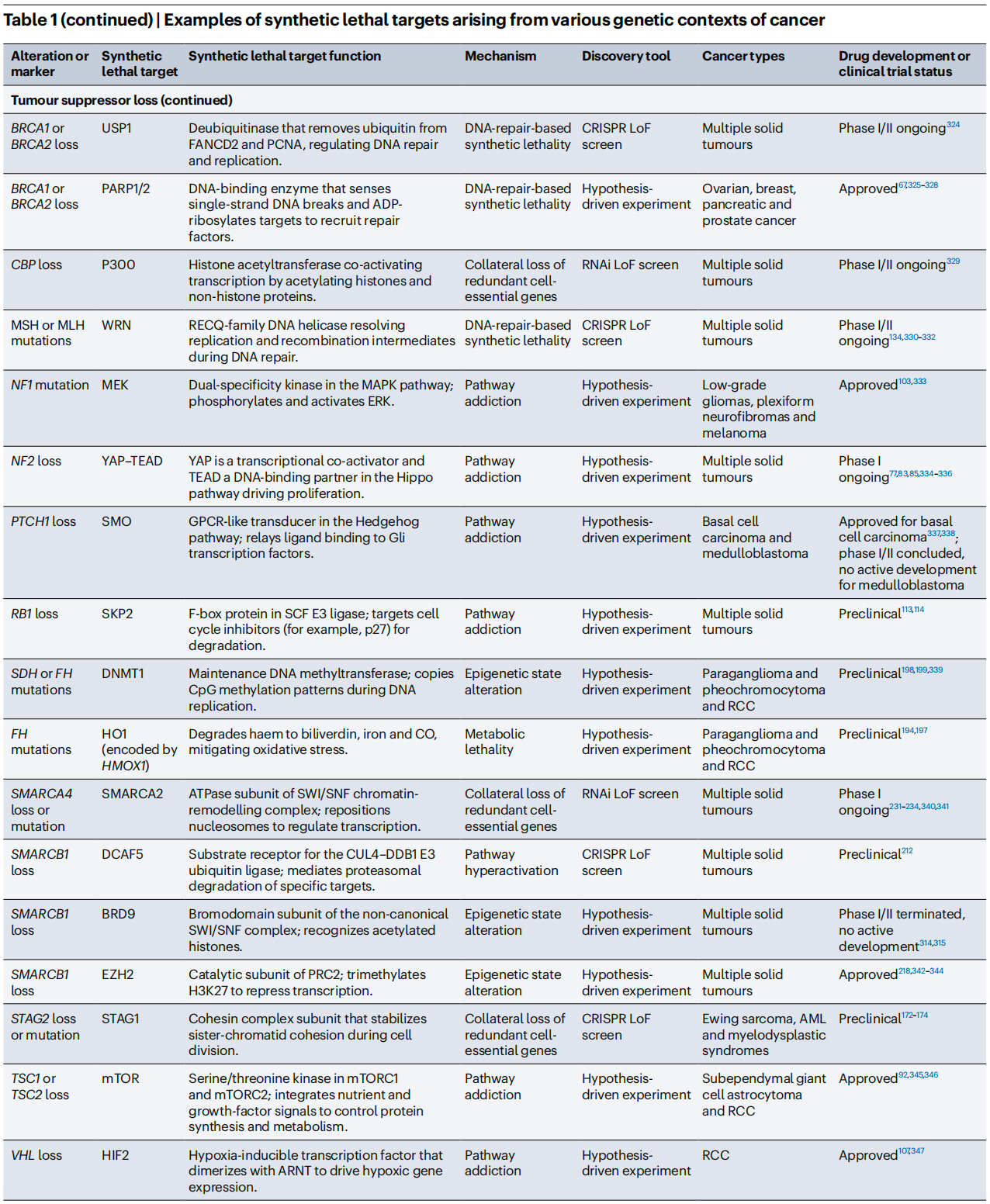
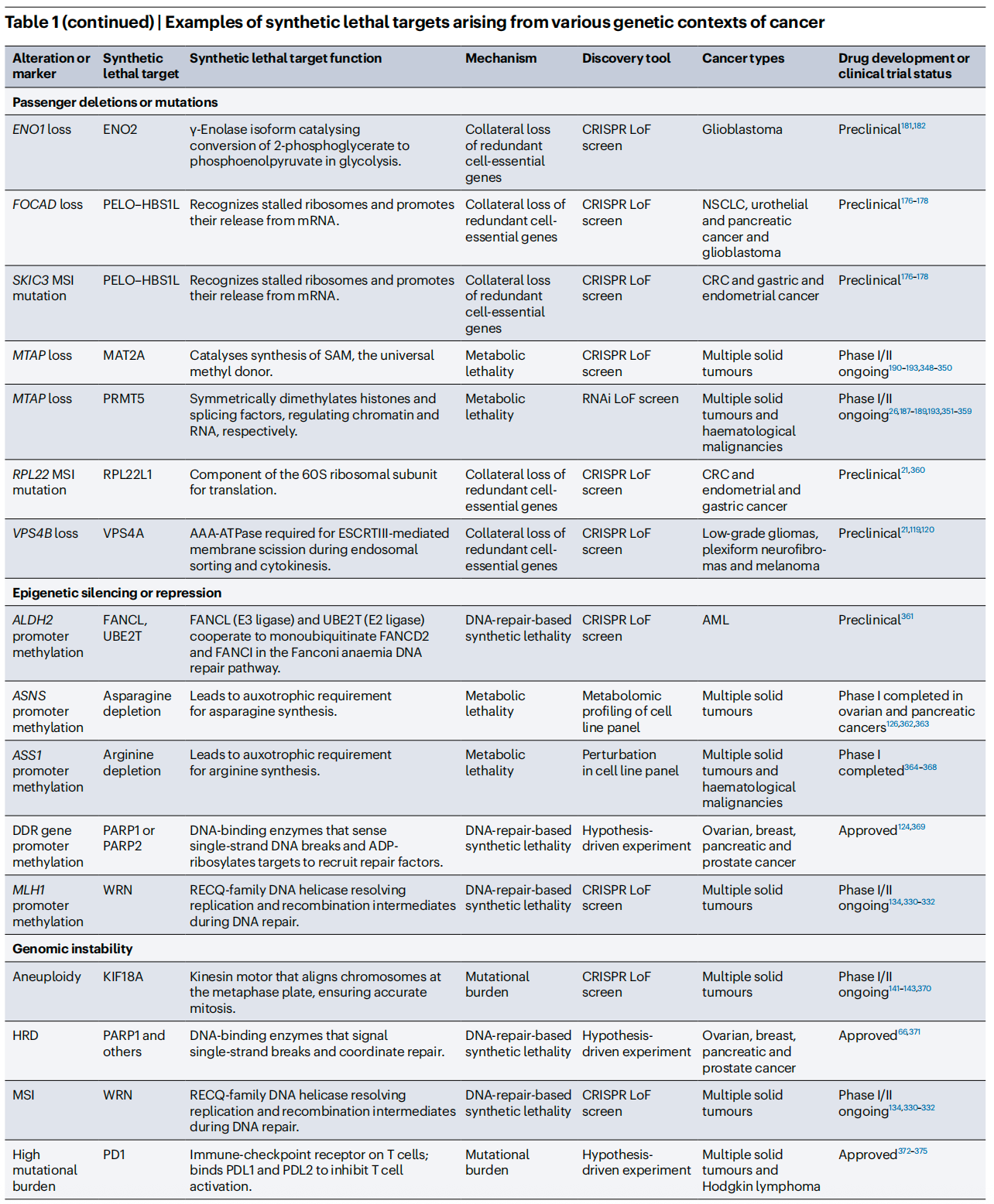

IV. From Bench to Bedside
Although PARP inhibitors have paved the way, not all synthetic lethality targets successfully translate into therapies. The review emphasizes two critical considerations:
- Therapeutic index is निर्णतive: The safety profile of targets varies significantly. Targets like PARP and WRN exhibit vulnerabilities highly restricted to biomarker-defined cancer cells, resulting in a wide therapeutic index and favorable safety. In contrast, targets such as WEE1 or ATR are broadly essential across many cell types, often leading to hematologic and gastrointestinal toxicities in clinical trials.
- Mechanism of action matters: Genetic knockout does not equal pharmacological inhibition. For example, in SMARCA4-mutant cancers, non-selective inhibitors cannot distinguish between normal and cancer cells. Targeted protein degraders (e.g., PROTACs) may provide a more effective and selective approach.
V. Discovering the Next Generation of Targets
Traditional single-gene knockout screens are reaching saturation. Future discovery strategies must evolve:
- Combinatorial genetic screening: Using CRISPR-based approaches to uncover hidden dual dependencies (e.g., DUSP4/DUSP6).
- Protein-level precision editing: Moving beyond gene knockout to identify vulnerabilities at the level of individual amino acids and protein domains.
- AI and machine learning: Leveraging large-scale datasets (such as DepMap) to uncover novel synthetic lethal interactions that may be missed by conventional statistical approaches.
VI. Conclusion
Synthetic lethality is pushing precision oncology into a deeper and broader dimension. By integrating genetic context with detailed molecular mechanisms—and combining these insights with emerging therapeutic modalities such as targeted protein degradation and allosteric inhibition—more effective and safer anticancer therapies are expected to move from the laboratory to the clinic in the years ahead.